3 Minutes
Cells depend on mitochondria for energy, but new research links mitochondrial errors in DNA replication to chronic inflammation that emerges with aging. Scientists from the Max Planck Institute for Biology of Ageing and collaborators examined human tissues and aging mouse models to identify a molecular mechanism that causes mitochondria to expel fragments of their own DNA into the cell, triggering inflammatory responses.
Scientific background: mtDNA, nucleotides and senescence
Mitochondria contain their own genome—mtDNA—which must be accurately copied during mitochondrial maintenance and replication. Nuclear DNA uses deoxyribonucleotides (dNTPs) as the building blocks for replication. Prior studies have noted that dNTP pools decline in aged and senescent cells, potentially compromising DNA synthesis.
When deoxyribonucleotides become scarce, mitochondrial polymerases can mistakenly incorporate ribonucleotides (rNTPs) instead. Ribonucleotide incorporation destabilizes DNA structure, creating lesions or imperfect copies that the organelle may detect as faulty and subsequently eject into the cytoplasm. Cytosolic mtDNA is recognized by innate immune sensors, provoking inflammatory signaling that can contribute to tissue damage.
Study design and key findings
Researchers used a combination of human tissue samples and genetically engineered mouse models that mimic age-related decline to trace how altered nucleotide balance affects mtDNA integrity and cellular inflammation. Through biochemical assays, sequencing and histological analysis, the team observed higher rates of ribonucleotide incorporation into mtDNA in aged or nucleotide-depleted tissues. These imperfect mtDNA molecules were more prone to being expelled from mitochondria.
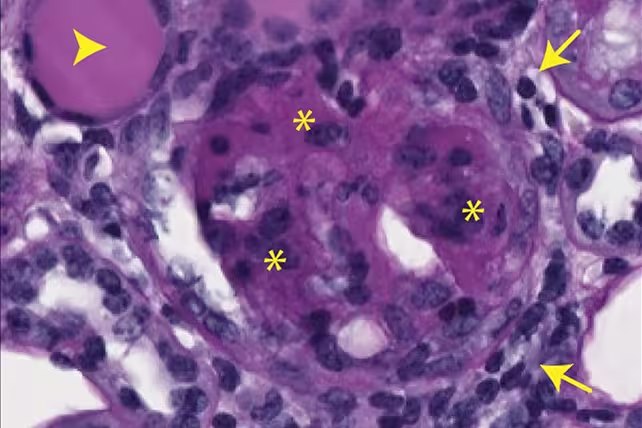
Evidence of kidney scarring in mice, caused by expelled mtDNA. (Max Planck Institute for Biology of Ageing)
The expelled mtDNA activated immune pathways associated with chronic inflammation and tissue scarring in animal models. The study connects metabolic changes—specifically dwindling deoxyribonucleotide availability—with the molecular chain of events that links mitochondrial dysfunction to inflammatory phenotypes in aging tissues.
Notable statements from investigators
Thomas Langer of the Max Planck Institute for Biology of Ageing summarizes the implication: metabolic disturbances that alter nucleotide pools can initiate a cascade leading to inflammation in senescent cells and aged organs, suggesting molecular targets for intervention. Dusanka Milenkovic, also at the institute, highlights that therapies currently used for some mitochondrial disorders—administering DNA precursors—might be worth testing to see whether they also reduce age-associated inflammation.
Implications for health and future research
This mechanism offers a plausible explanation for how low-level chronic inflammation builds up over decades and contributes to common age-related conditions, including certain cancers and neurodegenerative diseases such as Alzheimer’s. If scientists can prevent ribonucleotide misincorporation or stabilize mtDNA so it is not expelled, it may be possible to reduce damaging inflammatory signaling in older tissue.
Key next steps include determining how frequently this pathway operates during normal human aging versus in specific disease states, and whether therapies that restore deoxyribonucleotide pools can safely reduce mtDNA-driven inflammation without unintended effects.
Conclusion
The study links a metabolic shortfall—the reduction of deoxyribonucleotides—with a clear molecular route to inflammation via mtDNA instability and ejection. These findings open avenues for targeted strategies to preserve mitochondrial genome integrity and potentially curb inflammation-related decline in aging populations. Further preclinical and clinical work will be needed to evaluate whether nucleotide supplementation or other approaches can translate into measurable health benefits for older adults.
Comments
No comments yet.
Leave a Comment