6 Minutes
Summary and key finding
An international research team has demonstrated that increasing the activity of mitochondria — the cellular 'power stations' — can reverse memory and motor deficits in mice engineered to display dementia-like symptoms. Published in Nature Neuroscience, the study uses a research tool called mitoDREADD-Gs to selectively stimulate mitochondrial function in neurons, providing evidence that impaired mitochondrial activity can be a primary driver of neurodegenerative symptoms rather than only a downstream consequence.
The experiments show not just correlation but a causal relationship: when mitochondrial activity is chemically boosted, cognitive and motor impairments improve; when mitochondrial function is pharmacologically suppressed and then reactivated with the same tool, the deficits return and can be rescued again. These results point to mitochondria as a promising target for future therapies aimed at Alzheimer’s disease and related neurodegenerative disorders.
Scientific background: mitochondria and neurodegeneration
Mitochondria are intracellular organelles responsible for producing adenosine triphosphate (ATP), the energy currency that powers cellular processes. In neurons, which have high energy demands, mitochondrial performance is especially critical for synaptic signaling, plasticity, and survival. Over the past decade, many studies have associated mitochondrial dysfunction with aging and neurodegenerative diseases such as Alzheimer’s and Parkinson’s, but a persistent question has been whether mitochondrial failure is a cause of neuronal loss and cognitive decline or a secondary consequence of other pathological processes.
By creating an experimental framework to turn mitochondrial activity on and off in living neural tissue, the research team directly addressed that cause-and-effect problem. Giovanni Marsicano, a neuroscientist at INSERM, explains that the work is the first clear demonstration linking mitochondrial impairment to behavioral symptoms of neurodegeneration, suggesting that energy failure in neurons may initiate the cascade that leads to degeneration.
Experiment details: mitoDREADD-Gs, CNO, mouse models and human cells
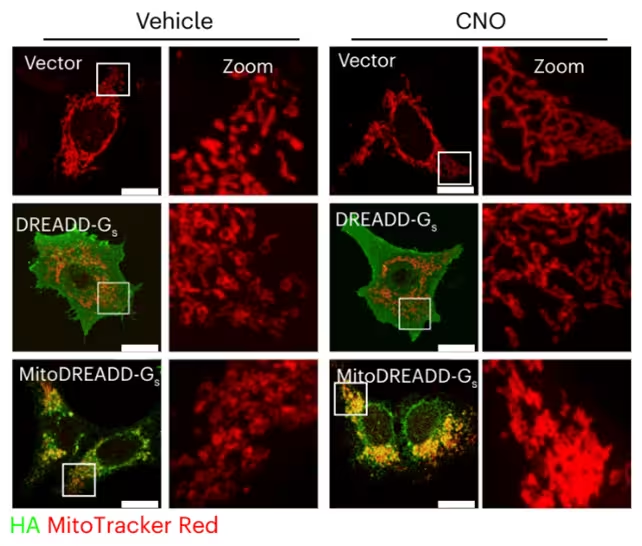
The core experimental advance is mitoDREADD-Gs, a genetically encoded actuator targeted to mitochondria that responds to a designer drug, clozapine-N-oxide (CNO). In effect, CNO acts as an ignition switch: binding to mitoDREADD-Gs increases mitochondrial signaling and stimulates ATP production in cells that express the actuator. With CNO treatment, mitochondrial activity (in red) was boosted (right-hand panels). (Zottola et al., Nat. Neurosci., 2025)
Researchers introduced mitoDREADD-Gs into neurons of mice genetically modified to model key features of dementia. They also tested the system in human cells cultured in the laboratory. In both settings, transient activation of mitochondrial activity improved memory performance in behavioral assays and ameliorated motor deficits. To test reversibility, the team used drugs to diminish mitochondrial activity pharmacologically; applying CNO to activate mitoDREADD-Gs then restored function, reinforcing the interpretation that mitochondrial impairment was responsible for the observed symptoms rather than incidental damage.
The experiments combined molecular biology, live-cell imaging, electrophysiology and behavioral testing to link cellular bioenergetics to organism-level outcomes. Importantly, mitoDREADD-Gs is a research tool rather than a therapeutic product. However, its ability to pinpoint when and where mitochondrial activation affects neuronal function provides a blueprint for identifying druggable molecular targets.
Implications for Alzheimer’s and future therapeutic strategies
The study suggests a new angle for developing treatments for neurodegenerative disease: restoring or sustaining mitochondrial performance in vulnerable neurons. Étienne Hébert Chatelain of the Université de Moncton notes that the tool can help map the molecular and cellular pathways that lead from energy failure to neuronal dysfunction, thereby guiding the search for effective therapeutic targets. If impaired mitochondrial activity is an upstream trigger in some forms of dementia, then pharmacological strategies that enhance mitochondrial metabolism or prevent its decline could slow or reverse symptoms.
Researchers emphasize several caveats. Dementia is heterogeneous: many genetic, metabolic, vascular, and environmental risk factors contribute to disease onset and progression. What works in a specific mouse model or a controlled cell culture system may not translate directly to the complex human brain. Long-term effects also require careful evaluation: chronic stimulation of mitochondrial function could have unanticipated consequences, including altered cellular signaling, oxidative stress, or metabolic imbalance. Luigi Bellocchio from INSERM highlights the next experimental priorities: measuring the effects of prolonged mitochondrial stimulation on neuronal survival, disease progression, and whether early intervention can delay or prevent neuronal loss.
Related technologies and complementary approaches
This work sits alongside other mitochondrial-targeted strategies under investigation, including small molecules that promote mitochondrial biogenesis, antioxidants that reduce reactive oxygen species, and gene therapies that correct mitochondrial DNA defects. Combining mitochondrial activation with treatments targeting amyloid, tau, inflammation or vascular health may be required to address the multifactorial nature of Alzheimer’s disease effectively.
Expert Insight
Dr. Maria Alvarez, a neuroscientist focused on cellular bioenergetics, commented: "These experiments provide compelling causal evidence that neuronal energy failure can drive behavioral symptoms. The mitoDREADD-Gs approach is an elegant way to control mitochondrial activity with temporal and spatial precision. For translation, the challenge will be to find clinically safe pharmacology that mimics this selective activation without provoking off-target effects."
Dr. Alvarez adds that future studies should prioritize longitudinal work in aged models and explore combinations with synapse-protective therapies to assess whether mitochondrial support can preserve neural circuits over months or years rather than hours or days.
Next steps and future prospects
The authors propose several follow-up directions: extending tests to other models of neurodegeneration and psychiatric conditions linked to mitochondrial dysfunction; mapping the downstream molecular pathways triggered by restored mitochondrial activity; and screening for drug candidates that can replicate mitoDREADD-Gs effects in human-compatible formulations. The translational path will require robust safety profiling, optimization of delivery mechanisms, and trials in large animal models before considering human clinical studies.
If mitochondrial failure proves to be a critical early event in certain forms of dementia, targeting cellular energy metabolism may become an essential part of a multi-pronged therapeutic strategy. Even if mitochondrial dysfunction is only one contributor among many, interventions that improve neuronal energy supply may slow symptom progression or enhance the efficacy of other treatments.
The research was published in Nature Neuroscience, and the experimental tool and data provide a roadmap for teams investigating mitochondrial roles in brain aging and disease.
Conclusion
This study offers strong experimental evidence that directly boosting mitochondrial activity in neurons can reverse memory and motor deficits in mouse models of dementia, supporting a causal link between mitochondrial dysfunction and neurodegenerative symptoms. While mitoDREADD-Gs itself is a laboratory tool rather than a clinical therapy, it reveals new molecular targets and experimental approaches that could inform future drug development. Critical next steps include assessing long-term effects, testing across diverse disease models, and identifying safe, human-compatible strategies to modulate mitochondrial function as part of comprehensive efforts to prevent or treat Alzheimer’s and related disorders.
Comments
No comments yet.
Leave a Comment